近日,我院2016级研究生朱艳迪同学在何豪副教授和李顺方教授以及其他老师的共同指导下在无缺陷衬底上高效单原子催化剂设计方面取得新进展,该工作以“Strain Engineering of Defect-free, Single-Layer MoS2 substrate for Highly Efficient Single-Atom Catalysis of CO Oxidation”为题发表在国际著名期刊ACS Appl. Mater & Inter.(影响因子:8.456)。见Yandi Zhu, Ke Zhao, Jinlei Shi, Xiaoyan Ren, Xingju Zhao, Yuan Shang, Xinlian Xue, Haizhong Guo, Xiangmei Duan, Hao He*, Zhengxiao Guo, and Shunfang Li*, ACS Appl. Mater & Inter. 1136,32887-32894 (2019) 文章链接:https://pubs.acs.org/doi/10.1021/acsami.9b06435。
单原子催化剂(SACs),由于其金属原子利用率高、具有单一的活性中心、催化选择性好等显著优点,已经成为近年来催化领域的研究热点课题之一。然而,严重限制SACs实际应用的瓶颈问题之一是SAC的不稳定性:在化学反应过程中容易在衬底上发生烧结或聚集成纳米团簇/纳米颗粒(NCs/NPs)。通常而言,为了阻止SACs烧结,人们在制备SACs时多时采用把昂贵的贵金属和过渡族金属原子钉扎在衬底表面缺陷位上。但是,衬底表面的本征缺陷的数量往往是有限的,同时,人为地制造大量的热力学稳定的表面缺陷也是十分困难。这几种因素严重限制了SACs的覆盖度,进而限制了SAC的稳定性、制备成本和催化效率。因此,寻求一种经济有效的方法提高在无缺陷衬底表面上SACs的稳定性和覆盖度是亟待解决一个重要科学问题。
针对上述问题,在何豪副教授和李顺方教授等老师的指导下,朱艳迪同学采用第一性原理计算方法,以二维材料MoS2为典型例子,研究了应力调控对无缺陷衬底的电子结构、物理化学性质的影响、特别是对衬底与沉积在其上的贵金属和过渡族金属的电子相互作用(EMSI)规律,进而研究实现高覆盖度和高效催化活性的SAC的可行方案和物理机制。选取二维材料的主要原因如下:1) 二维材料的表面积/体积比例更高,活性往往更高; 2)表面稳定吸附位更多;3)相对于三维块体材料而言,二维材料的结构和属性更容易被调控。
首先,该研究以两个贵金属Pd原子在无缺陷的单层MoS2衬底(简称P-MoS2)上的相互作用作为典型例子,来揭示调控SAC稳定性的物理机制。研究发现,在P-MoS2衬底上,Pd原子更倾向于二聚体,即Pd-SAC并不稳定;然而拉应力可以有效增强EMSI,即增加Pd原子和衬底之间的电荷转移和轨道杂化并形成更强的离子键,同时大大降低Pd原子在衬底表面上的扩散系数,从而有利于 Pd2二聚体解离成稳定的Pd-SAC, 并对CO氧化表现出高效可控的催化性能 (见图1)。 其次,这种简单有效的应力调控手段被证实同样适用于很多更加廉价的过渡金属元素(比如:Ag, Ni, Cu, 和Cr)。这些元素都可以通过应力调控实现从团簇到单原子相的转变,并对CO氧化具有高效催化作用。另外,该研究还建立一套简单有效的判据来判断一个无缺陷二维材料上SAC能否稳定(不团聚),该判据已经被最近发表的实验文章所支持[Lang, et al., Nat. Commun.10, 234(2019)]。该研究为制备高覆盖度、稳定、经济、高效SACs提供了重要的新思路,为调控SAC的反应选择性提供了明确的理论指导。该工作得到了国家自然科学基金面上项目和物理学院物理学科提升计划的支持。
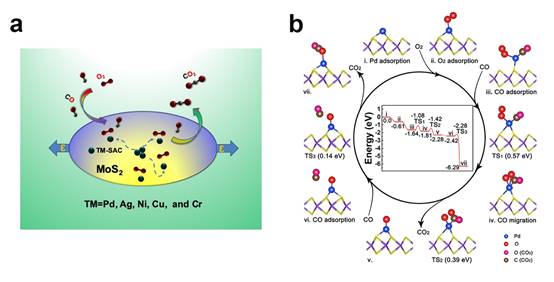
图1:(a)拉应力调控稳定的TM1-SAC示意图; (b)及Pd-SAC对CO催化氧化主要过程。