凝聚态计算与理论物理研究所硕士研究生赵珂同学在任晓燕老师和李顺方教授的指导下在高效单原子催化剂设计方面取得新进展,该工作近日以“Synergetic effects of strain engineering and substrate defects on generating highly efficient single-atom catalysts for CO oxidation”为题发表在国际知名期刊J. Mater. Chem. A上(影响因子:9.93)。见Ke Zhao, Yandi Zhu, Jinlei Shi, Xingju Zhao, Rui Pang, Xinlian Xue, Xiaoyan Ren*, Xiangmei Duan, Z. X. Guo, and Shunfang Li*, J. Mater. Chem. A, 2019, 7, 9297-9304,文章链接:
https://pubs.rsc.org/en/content/articlelanding/2019/ta/c9ta01326a#!divAbstract
近些年来,过渡族金属和贵金属单原子催化(SAC)(即以单个原子形式高度分散在合适的衬底上的单原子催化剂体系)的制备、表征、催化机制的理解以及催化性能调控已经成为多相催化领域的前沿热点课题。然而,研究发现,SACs在化学反应过程中往往会成核长大为团簇或烧结为纳米颗粒,从而降低催化效率和化学反应的选择性,严重阻碍了其应用和发展。因此,理解单原子在衬底上的成核物理规律以及如何抑制其成核已经成为显著增加单原子催化剂覆盖率和催化效率的关键科学问题。
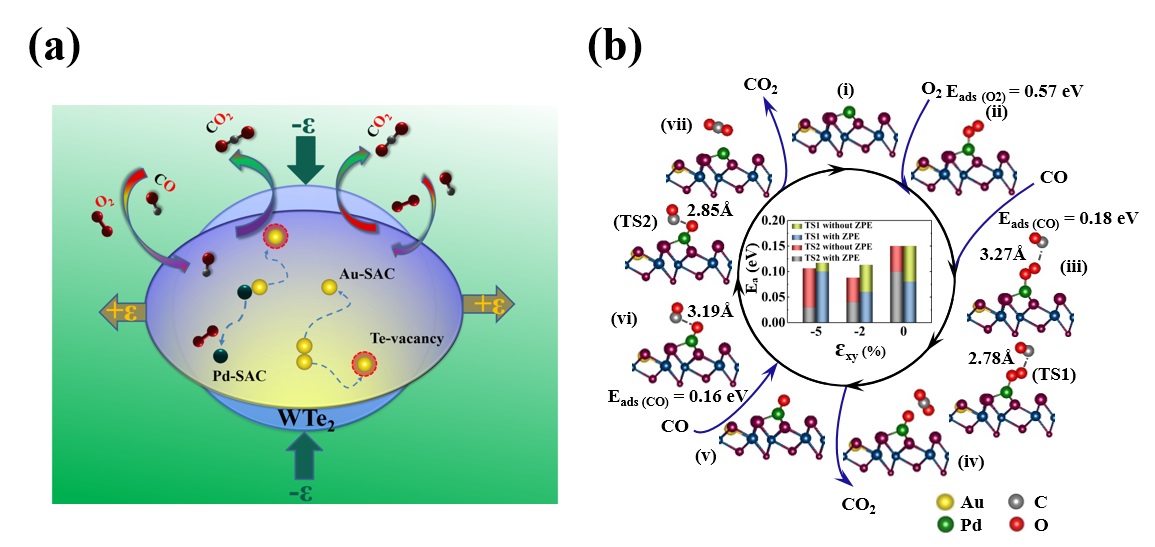
针对上述问题,任晓燕老师和李顺方教授指导硕士研究生赵珂同学采用基于密度泛函理论的第一性原理计算方法,以沉积到单层WTe2表面上的电子闭壳层Au2和开壳层Pd2 (PdAu)贵金属体系作为最小团簇的典型例子,通过应力和衬底缺陷的协同效应来调制金属原子-衬底之间相互作用(EMSI),对SACs成核的热力学和动力学性质进行了深入研究。研究结果表明,在完整的WTe2(P-WTe2)衬底上,Au和Pd原子都倾向于成核为二聚体而不是互相分离;但是,当同一个WTe2衬底上存在Te原子缺陷(D-WTe2)时,这些贵金属原子的行为发生了很大变化,衬底缺陷具有稳定单原子相的趋势。在对衬底施加外应力时,相对于具有电子闭壳层结构的Au2二聚体,具有电子开壳层的单个Au原子占据Te空位处(VTe)时,后者从WTe2衬底上获得更多电荷,从而产生更强的EMSI。然而,对于具有电子开壳层的PdAu(Pd2)二聚体而言,对衬底施加压应力时,金属原子之间的间距缩短,同时更多的电荷转移到两个沉积原子上,因此增加的库仑排斥作用将它们分离成稳定的SACs(示意图(a))。重要的是,我们发现应力可以有效的放大缺陷对单原子的钉扎效应,从而使这些昂贵的贵金属单原子在衬底上的结合能显著增强,难以扩散,从而避免单原子之间相互碰撞成核的几率。并且,应力可以对这些单原子对CO催化氧化势垒进一步所调控,如图(b)所示。该理论计算工作预言了一种简单有效的方法,即衬底应力工程来稳定并显著提高单原子催化剂的覆盖度,从而显著提高其催化效率和选择性。
文章投稿后,受到两个审稿人高度评价。Referee 1: “This is an interesting computational work towards to design catalysts for CO oxidation; it is valuable and self-supported…”; Referee 2: “Overall, this is a well-conceived and carefully performed project, and the results may be further extended to related system.” 该工作已经受到国内多个实验课题组的关注。