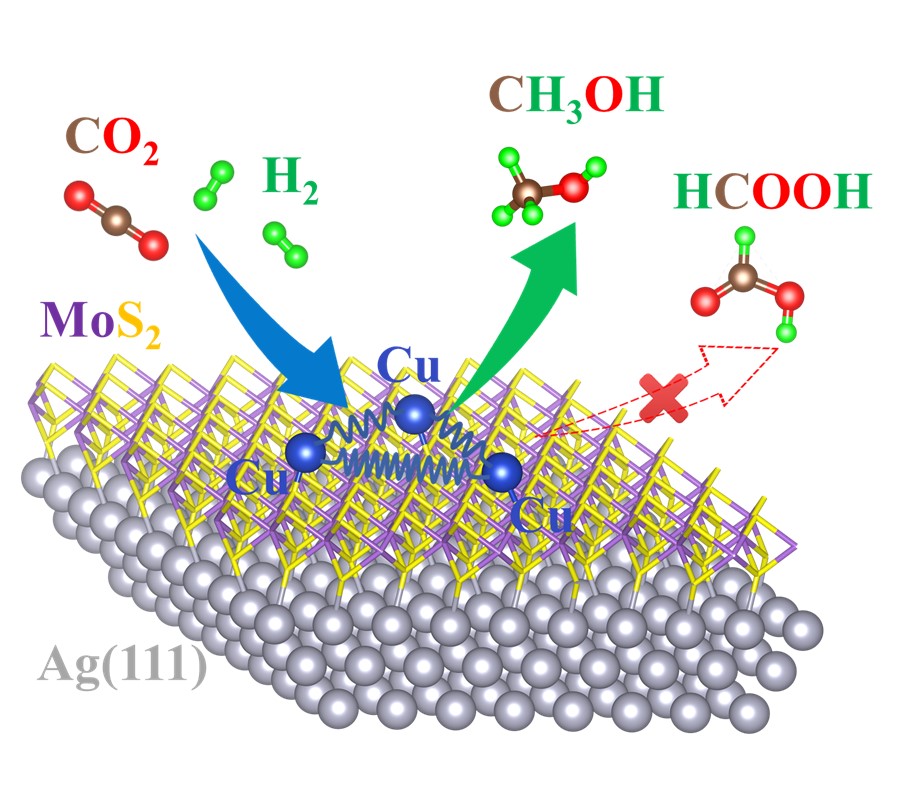
近日,郑州大学物理学院凝聚态计算与理论物理研究所李顺方教授团队在单原子尺度团簇选择性催化CO2制CH3OH方面取得重要新进展,相关成果以《Selective Hydrogenation of CO2 to CH3OH on a Dynamically Magic Single-Cluster Catalyst: Cu3/MoS2/Ag(111)》为题,在线发表在催化领域国际顶级期刊《ACS Catalysis》上。论文第一作者和共同第一作者分别为我院硕士研究生王亚婉和博士研究生朱艳迪同学,本科毕业生朱晓雯、史金磊博士后、任晓燕副教授为共同作者,通讯作者为我院青年教师张丽丽博士和李顺方教授。全文链接:https://pubs.acs.org/doi/10.1021/acscatal.2c05072。
随着能源危机和环境污染、温室效应的加剧,国际社会达成碳达峰和碳中和的一致目标。 国家主席习近平在第七十五届联合国大会上宣布,中国力争 2030年前CO2排放达到峰值,努力争取 2060年前实现碳中和目标。多年来,人们致力于捕获CO2并将其还原转化为具有高附加值的碳氢化合物,该项措施是实现上述宏伟目标的重要途径之一。 而寻找高效经济的催化剂成为CO2催化转化中的关键因素。相对于其它昂贵的贵金属催化剂体系,Cu基催化剂对CO2的催还还原表现更为优异:Cu基催化剂被发现是目前为数不多的、能够高效实现多种还原产物的候选材料。在众多CO2还原生成的产物中,甲醇(CH3OH)不仅具有清洁燃烧、高生物降解性的特点,同时是一些高价值下游产品的关键原料,因而备受关注。因此,寻找经济高效的单原子尺度催化剂、厘清其催化转化CO2的微观物理机制、降低CO2还原生成CH3OH的成本、并提升其催化反应的选择性,一直以来都是一个具有挑战性的研究课题。
该团队以沉积在MoS2/Ag(111)异质结沉底上的CuN团簇,即CuN/MoS2/Ag(111)体系为模型催化剂(N=1~8),从第一性原理计算的角度揭示了单原子尺度Cu基催化剂活性位点CuN的“动态幻数”特性在还原CO2转化为CH3OH过程中的重要作用。研究发现:在气相下,Cu2和Cu8因其闭壳层的电子组态而展现出超高的稳定性和化学惰性,即“幻数”特征;然而,当CuN沉积于MoS2/Ag(111)衬底上之后,由于强的金属-衬底相互作用和电荷转移(Cu-S离子键),Cu3和Cu7的价电子数最接近闭壳层,成为新的幻数结构。然而,局域在Cu3团簇的s-d杂化轨道的电子有点类似于碱金属原子的电子壳层排列形式,从而容易“捐赠”部分电子到CO2的LUMO轨道,且其原子结构在催化反应中呈现出明显的动态演化的特点,从而显著降低反应过程中决速步的势垒,仅仅约为1.10 eV,明显低于最近所报道的其他Cu基高效催化剂体系(~1.4 eV); 另外,对CO2选择性加氢还原为CH3OH而非HCOO产物表现出很高的催化选择性性。然而,沉积的Cu7的电子组态类似于卤族元素的电子结构,不易捐赠电子给CO2,从而对CO2的活化表现出很高的化学惰性。该研究结果对设计和制备高效经济的单原子尺度催化剂实现CO2的还原转化具有重要理论指导意义。
该工作得到了国家自然科学基金的支持。本文的计算是在国家超级计算郑州中心完成的。