河南省量子功能材料国际联合实验室黄扬同学在王飞老师和贾瑜教授的指导下在第一性原理计算结合机器学习预测材料性质方面取得新进展,该工作近日以“Band gap and band alignment prediction of nitride-based semiconductors using machine learning”为题发表在国际知名期刊J. Mater. Chem. C上。见J. Mater. Chem. C, 2019, 7, 3238-3245,文章链接:https://pubs.rsc.org/en/Content/ArticleLanding/2019/TC/C8TC05554H
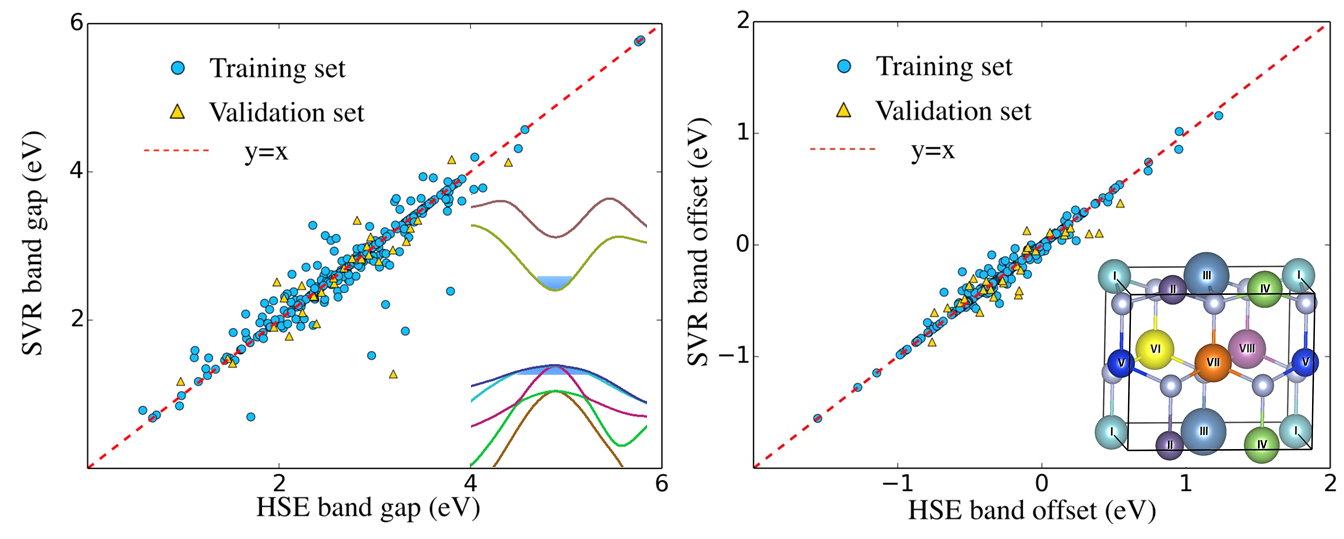
近年来,机器学习在新材料的发现和设计中发挥着越来越重要的作用。王飞老师,贾瑜教授和信息工程学院刘玉怀教授合作,指导研究生黄扬同学采用基于密度泛函理论的第一性原理计算方法结合机器学习算法,设计预测了GaN基氮化物材料的带隙和带阶性质,为新材料的发现和设计提供理论指导。他们首先根据价电子数目设计了一大类氮化物,随机选取其中的300个为样本,用第一性原理PBE和HSE势函数计算了它们的带隙和带阶。通过多种机器学习算法对比,发现支持向量回归(support vector regression, SVC)得到的带隙和带阶的均方根误差最小,分别为0.298 eV and 0.183 eV。特别的当把PBE计算的带隙也作为特征加入时,带隙的均方根误差进一步降低至0.1eV以下。并通过特征工程算法,分析了不同特征量对带隙的影响贡献。最后预测了设计空间中所有氮化物材料的带隙和带阶值。该工作开辟了实验室新的计算方法,并为实验制备相关氮化物提供了精确的理论指导。