1引言
锂离子电池(LIBs)作为应用最广泛的可持续能源器件,一直深受严重安全问题的困扰。此外,日益增长的社会能源需求也推动着从传统液态电解质到基于固态电解质的电池系统的革命性变革。与液态电解质相比,固态电解质具有更高的安全性、高能量密度和长寿命周期。其中,无机晶体固态电解质(ICSSEs),如Li6PS5Br和Li6PS5Cl(LPSC),相较于液态电解质具有更高的离子电导率,因而引起了广泛关注。然而,由于工作条件(包括温度和恒定电势)下多尺度锂离子传输过程固有的复杂性,原子尺度的结构缺陷以及复杂的相互作用之间的关系,种种因素导致LPSC电解质的内在机制仍难以阐述清楚。
2成果展示
近日,郑州大学化工学院周震教授团队的张旭和田芸教授在这一领域取得了关键进展。他们用经典分子动力学耦合恒电势的方法,首次在“恒定电压”条件下还原了锂离子在典型固态电解质Li6PS5Cl中的迁移全景,揭示了锂空位在锂离子传输过程中扮演的重要角色。
结果揭示了锂离子沿着空位的传输路径,并且得到了化学组成为Li5.3PS4.3Cl1.7的最优结构,首次实现了与实验数据的半定量吻合。该研究除了关注锂离子的传输路径、Cl/S组成比对LPSC型固态电解质性能的影响,还揭示了电场作用下,固态电解质的一些传质特性:1) 电解质与电极界面的电势分布呈现出振荡结构,与传统液态电解质体系差异巨大。2) 电场作用的存在虽然会影响锂离子的迁移速率,但是对于整体配位环境的改变忽略不计。整体而言,作者通过经典分子动力学模拟与恒定电势方法的结合,实现了对受限体系中离子传输行为的真实还原,从多尺度视角为固态电解质的理论建模弥合了关键鸿沟。其成果以题为“Unveiling Li-ions migration mechanism in Li6PS5Cl under applied constant potential: insights from classical molecular dynamics simulations”为题发表在期刊npj Computational Materials期刊上。
3图文导读
电池工作条件下固态电解质中锂离子多尺度传输现象的阐释,对实验和理论领域均构成巨大挑战。高分辨率第一性原理分子动力学模拟受限于时空尺度,难以与室温外加电势条件下的实验观测结果直接比对。本研究采用恒电位经典分子动力学模拟方法,在真实工况下揭示了电极界面约束的Li6PS5Cl(LPSC)中锂离子的迁移机制。通过对LPSC电解质中阴离子组成的精细调控,发现相邻空位为锂离子迁移提供了有效通道,且配位环境随着扩散系数的增加发生渐进式演变,而Li5.3PS4.3Cl1.7组分的电导率呈现非单调峰值特征。与现有实验结果的半定量吻合证明了本研究所构建恒电位固态电解质模型的优越性,该模型有望为固态电解质的理性设计提供原子尺度认知。
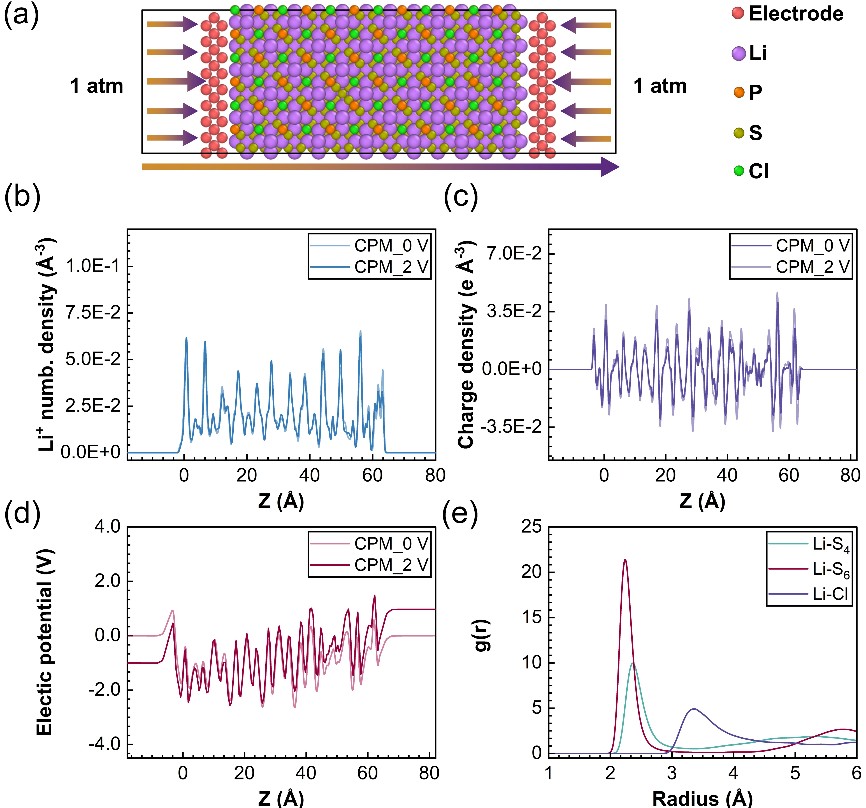
图1. LPSC 电解质模型快照:(a) 初始模型;(b) 锂离子浓度分布;(c) 电荷密度分布;(d) 电势分布;(e) 锂离子的径向分布函数。
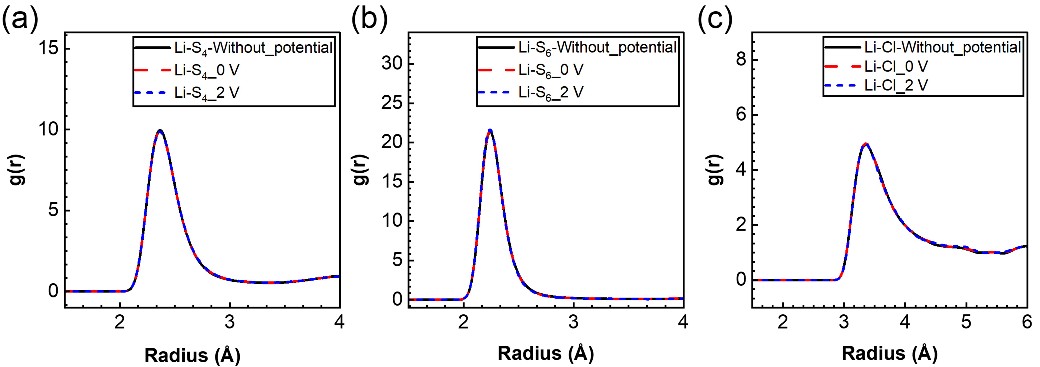
图2. 锂离子在不同电势下的径向分布函数
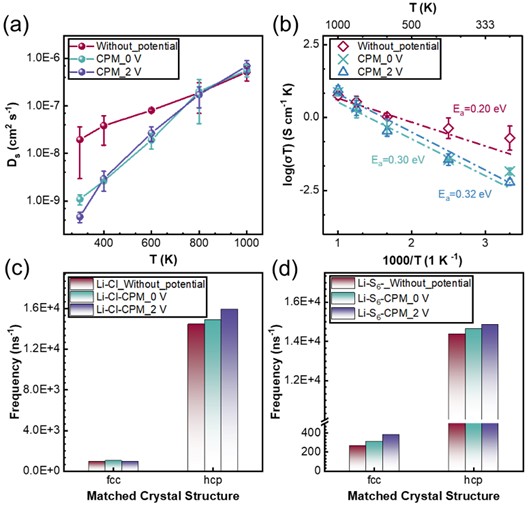
图3. 温度和电势对LPSC中锂离子迁移的影响:(a) 锂离子扩散速率随温度的变化;(b) 随温度变化的阿伦尼乌斯曲线; (c, d)锂离子在恒电势下与阴离子形成的空间结构。
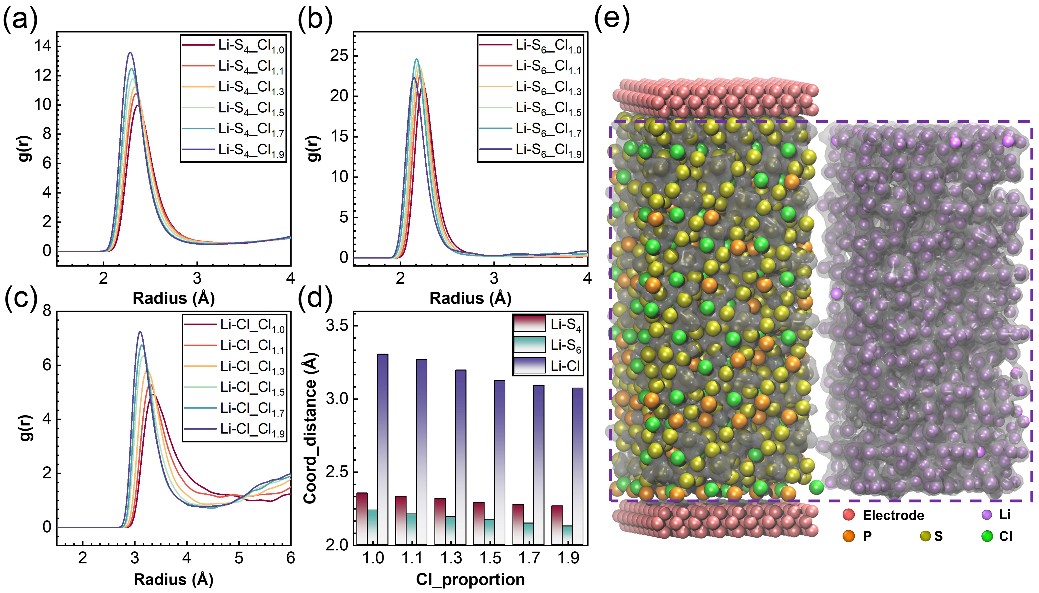
图 4. 锂离子在Li6-xPS5-xCl1+x中的配位环境:(a) Li-S4的径向分布函数;(b) Li-S6的径向分布函数;(c) Li-Cl的径向分布函数;(d) 平均配位距离的总结;(e) 锂离子概率密度分布。
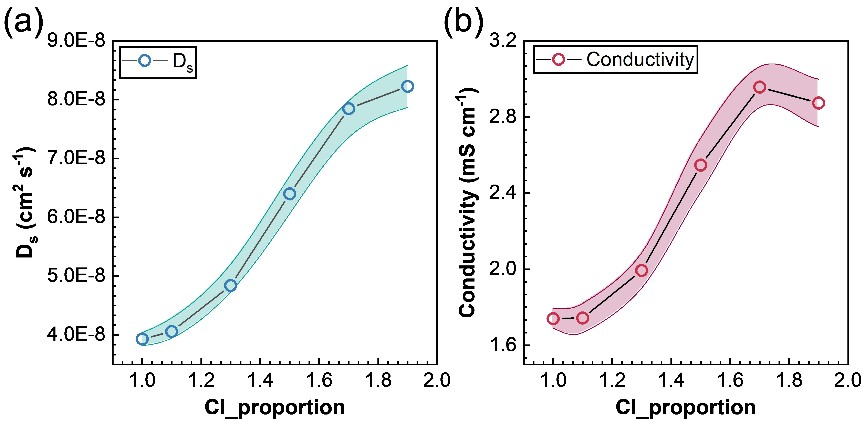
图5. Li6-xPS5-xCl1+x中的传质性能:(a) 扩散系数随Cl含量的变化;(b) 电导率随Cl含量的变化。
4总结
采用经典动力学方法研究了外加恒电势下锂离子在LPSC固态电解质中的传输机制。模拟结果表明,外加电场对Li+在传输过程中的配位环境影响甚微,而离子电导率随Cl组分的增加呈现非单调变化趋势,其中Li5.3PS4.3Cl1.7被具有最佳性能。发现还印证了,对于LPSC典型ICSSEs中,相邻空位是Li+迁移的主要通道。本研究为实际条件下固态电解质中的传质机制提供了有价值的见解,并且期望借此加速固态电池中ICSSEs的理性设计。此外,经典MD模拟与恒电位法(CPM)的结合,使得能够真实描述受限体系中的离子传输行为,从多尺度视角弥合了固态电解质理论建模的鸿沟。
文章信息
Unveiling Li-ions migration mechanism in Li6PS5Cl under applied constant potential: insights from classical molecular dynamics simulations
Mengwei Sun, Wenchuang Yuan, Xu Zhang*, Yun Tian*, Zhen Zhou
Npj Computational Materials
https://doi.org/10.1038/s41524-025-01832-x
作者信息

孙孟威 (第一作者): 2023年至今就读于郑州大学化工学院,化学工程专业,指导老师为田芸、张旭老师,主要研究为固态锂离子电池的动力学模拟和智能设计。

张旭 (通讯作者): 现为郑州大学化工学院直聘研究员,多年来致力于结合多尺度模拟、高通量筛选和机器学习等方法,围绕能量存储与转化材料的设计、筛选及界面催化反应机理的探索等方面开展系统性工作。以(共同)第一/通讯作者在Natl. Sci. Rev.、J. Am. Chem. Soc.、Angew. Chem. Int. Ed.等期刊发表论文50余篇,引用9000余次。

田芸 (通讯作者): 郑州大学化工学院副教授,2012年和2017年分别于华东理工大学和美国加州大学河滨分校获得工学学士和博士学位,博士期间师从统计热力学专家吴建中教授。主要研究方向是结合经典密度泛函理论等统计热力学方法与机器学习算法,对限域空间中流体在主体和界面相的热力学平衡分布和动力学迁移性质进行多尺度模拟,预测电池的宏观性能并进行系统优化。主持国家自然科学基金面上项目一项,国家自然科学基金青年项目以及河南省优秀青年基金。近年来以通讯作者或第一作者身份在AIChE J.、Angew. Chem. Int. Ed.、Adv Mater、Energy Environ Sci.、ACS Energy Lett.等期刊上发表论文近20篇。现为《化工学报》(英文版)青年编委。





