近日,郑州轻工业大学材料与化学工程学院巩飞龙博士、张永辉教授联合中国科学院大连化学物理研究所刘健研究员报道了一种通用的亚纳米反应器策略合成蛋黄蛋壳MoS2负载的单原子电催化剂。该研究成果的模拟计算部分得到了国家超级计算郑州中心“嵩山”超级计算机平台强大算力支持。相关研究成果以《Universal Sub-Nanoreactor Strategy for Synthesis of Yolk-Shell MoS2Supported Single Atom Electrocatalysts toward Robust Hydrogen Evolution Reaction》为题发表在国际权威期刊《Angewandte Chemie International Edition》上,论文入选Hot Topic,并被选为Back Cover封面文章刊出。
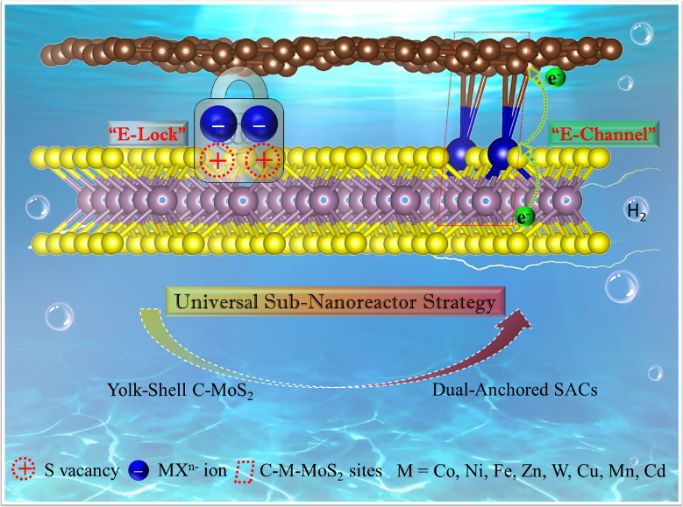
开发高效节能的电解水制绿氢技术对解决能源需求和实现“双碳”目标至关重要。单原子催化剂由于其高活性和最大化的原子利用效率受到广泛关注,然而,高表面能使得单原子在电催化环境中易聚集,影响催化效率和稳定性。调节单原子在碳基或非碳基载体上的配位结构可促进电催化选择性和活性,但精确合成具有空间位置以及构建兼具碳基和非碳基双配位微环境的单原子催化剂仍极具挑战。该工作提出一种通用的亚纳米反应器策略,将金属单原子限域在亚纳米空间内,精准构建系列兼具二硫化钼和插层碳双配位微环境的单原子催化剂,用于高效稳定电解水制氢,其中,C-Co-MoS2表现出优于迄今为止报道二硫化钼基单原子催化剂的产氢活性。
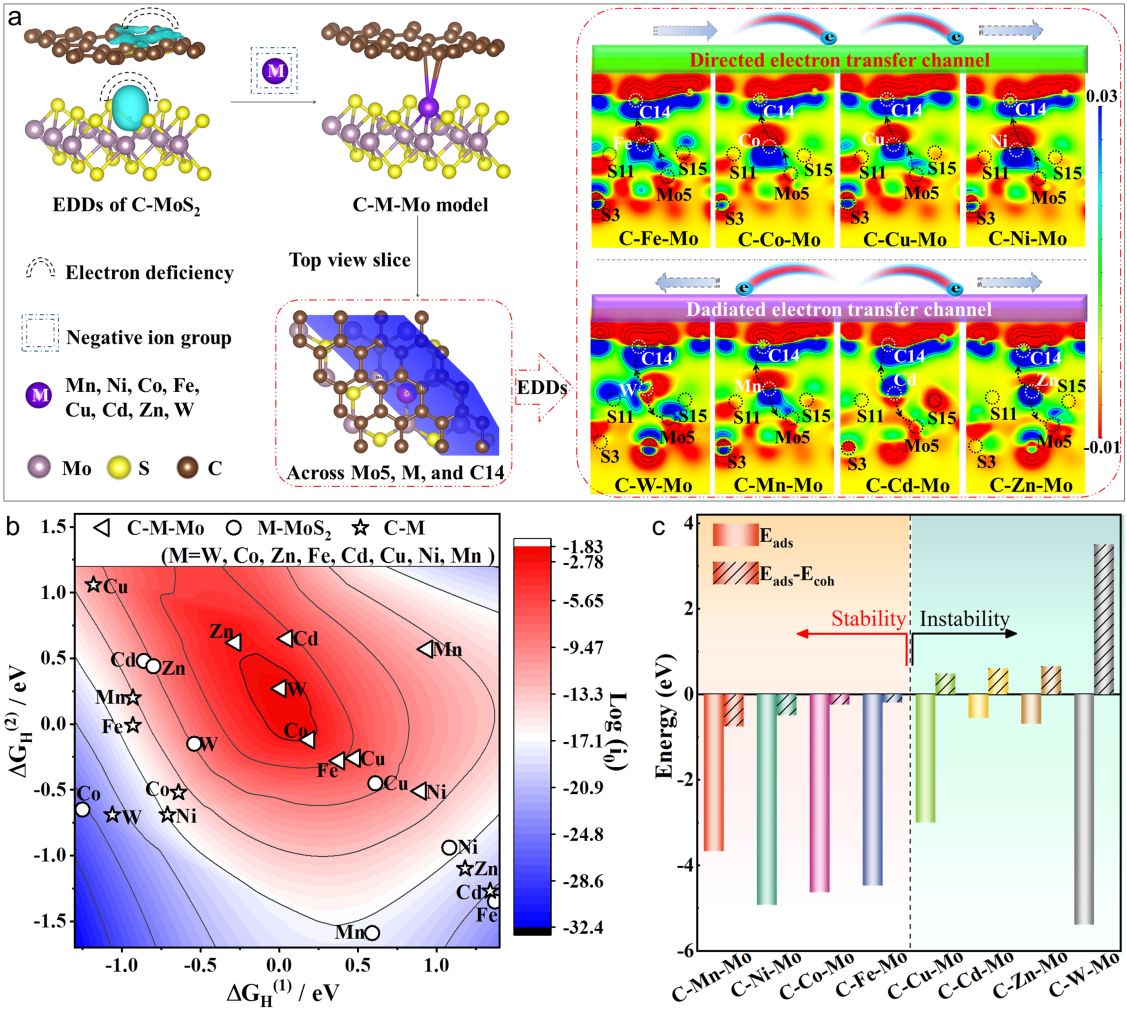
图1活性和稳定性理论计算。(a)C-MoS2的EDDs、C-M-Mo的结构模型、顶视图以及相应的EDDs;(b)基于ΔGH(1),ΔGH(2)和log (i0)建立的不同结构3D活性火山图;(c)C-M-Mo单原子模型的稳定性。
模拟计算部分以具有硫空位的MoS2和插层碳为基础,构建了系列单/双配位单原子催化剂模型,建立了由ΔGH(1)和ΔGH(2)、吸附能以及吸附能与内聚能差值决定的活性和稳定性描述符。根据电荷差分密度和相应的Bader电荷分析结果显示,金属单原子可以在硫空位和插层碳的共同作用下稳定在双载体中,同时在C-M-Mo界面上产生定向和发散的两种电荷转移通道。